Genomisches Monitoring — was ist das überhaupt und was hat es mit Naturschutz zu tun?
Genomic monitoring — what is that supposed to be, and what does it have to do with conservation?
Eine Herausforderung
„Biologische Vielfalt“ bezeichnet den Variantenreichtum unter den lebenden Organismen aller Herkünfte, einschließlich […] der Vielfalt innerhalb von Arten, der Vielfalt der Arten und der Vielfalt der Ökosysteme.
Übereinkunft zur Biologischen Vielfalt, Vereinte Nationen, 1992
Seit dem Jahr 1992 haben sich weltweit fast alle Staaten in einem internationalen Abkommen, der „Übereinkunft zur Biologischen Vielfalt“ (engl. „Convention on Biological Diversity“, kurz CBD), zum Schutz der biologischen Diversität sowie zu Nachhaltigkeit und Fairness bei deren Nutzung verpflichtet. Doch während man sich die Vielfalt der Ökosysteme – z.B. Meere, Wälder, Wiesen, Hochmoore … – und die der Arten – z.B. Arnika, Gänseblümchen, Honigbiene, Giraffe … – noch gut vorstellen und sie meist auch leicht beobachten und messen kann, stellt uns die „Vielfalt innerhalb von Arten“ vor Herausforderungen. Gemeint sind hier – nicht nur, aber vor allem – genetische Unterschiede zwischen Angehörigen der selben Art: Ein Konzept, das wir von uns selbst sowie von unseren Nutzpflanzen und -tieren sehr gut kennen, das aber auf sämtliche Arten der Erde zutrifft.
Manche genetischen Unterschiede sind nach außen sichtbar – beispielsweise Varianten in den Genen für Haar- und Augenfarbe bei Menschen oder Blütenfarbe und Fruchtform bei vielen Gartenpflanzen – während man andere dagegen nur entdeckt, wenn man die DNA-Sequenzen zweier Individuen direkt vergleicht. Wie aber soll man etwas schützen, das man gar nicht sieht – und zu dessen Veränderungen in der Natur man im Moment zwar viele Theorien, aber wenig Beobachtungen hat?
A challenge
„Biological diversity“ means the variability among living organisms from all sources including […] diversity within species, between species and of ecosystems.
Convention on Biological Diversity, United Nations, 1992
Since 1992, almost all states of the world have pledged themselves to protect biological diversity and to only use it in a sustainable and fair way, as set down in the international Convention on Biological Diversity (in short: CBD) of the United Nations. Yet while we can easily picture, and usually also observe and measure, the diversity of ecosystems – e.g. oceans, forests, meadows, bogs … – and the diversity between species (or species diversity) – e.g. Arnica, daisy, honeybee, giraffe … – „diversity within species“ poses some challenges. What is meant here, are – usually, but not exclusively – genetic differences between members of the same species: a concept we know well from our own species and those we cultivate or breed, but which equally applies to all species on our planet.
Some genetic differences are visible on the outside – e.g. hair and eye colors in humans, or flower color and fruit shape in many garden plants – but others can only be noticed through a direct comparison of the DNA sequence of two individuals. But how are we to protect something which we cannot see – and about the natural dynamics of which we currently have more theories than observational data?


Neue Technik
Leider gibt es bis heute (noch?) keine Technologie, mit der wir DNA-Sequenzen von Organismen ganz einfach lesen können, indem wir nur vor ihnen sitzen. DNA-Sequenzierung bedeutet zunächst (meist) mehrere Tage Arbeit im Labor, den Einsatz von Spezialgeräten und -chemikalien sowie anschließend Rechenarbeit am Computer – je nach Menge der Daten inzwischen häufig unter Einsatz eines Hochleistungsrechners. Im Lauf der Zeit wurden deshalb verschiedene Verfahren entwickelt, um den Aufwand zu reduzieren und gleichzeitig immer genauere und vollständigere Daten zu eventuellen Unterschieden in der Genom-Sequenz von Individuen zu bekommen.
Mikrosatelliten / SSR (für engl. „short sequence repeats“) sind Bereiche im Genom, an denen das immer gleiche, kurze Sequenzmotiv (z.B. „AC“, „GTT“ oder ähnliche) mehrfach hinter einander wiederholt wird (z.B. „ACACACACAC“, „GTTGTTGTT“). Beim Vervielfältigen der DNA während der Zellteilung bekommt das dafür zuständige Enzym, genannt DNA-Polymerase, hier häufig „Schluckauf“ und verzählt sich. Daraus ergeben sich Unterschiede in der Anzahl der Kopien des Sequenzmotivs, bzw. in der Länge des gesamten Mikrosatelliten-Bereichs. Sind die „normalen“ Sequenzen vor und nach dem Mikrosatelliten-Bereich bekannt, kann man diesen im Labor gezielt kopieren, seine Länge bestimmen – und zwischen verschiedenen Individuen vergleichen. Allerdings ist diese Methode fehleranfällig: Varianten mit gleicher Länge / Kopienanzahl können mehrfach auf verschiedenem Weg entstanden sein, auch andere Mutationen können die Länge eines Mikrosatelliten-Bereiches verändern und manchmal gibt es den scheinbar selben Mikrosatelliten-Bereich gleich an mehreren Stellen im Genom, sodass man unter Umständen Äpfel mit Birnen vergleicht.
AFLP (für engl. „amplified fragment lenght polymorphisms“) vergleichen ebenfalls nur die Länge und nicht die Sequenz von bestimmten Bereichen im Genom. Dazu wird die gesamte DNA eines Individuums mit einer anderen Sorte von Enzym, sogenannten Restriktionsenzymen, die den DNA-Doppelstrang an charakteristischen Sequenzmotiven (z.B. „A|CCG“) schneiden, in kurze Schnipsel zerlegt und deren Länge gemessen. Bei genetisch identischen Proben würde man dann erwarten, dass auch die Liste der so erzeugten Fragmentlängen identisch ist – anderenfalls nicht. Allerdings leidet auch dieses Verfahren unter den selben Einschränkungen wie die Längenvergleiche der SSRs, durch die Verarbeitung des gesamten Genoms sogar in noch stärkerem Maß.
Seit einigen Jahrzehnten gibt es ein neues Sequenzierverfahren (Illumina-Sequenzierung, nach der Herstellerfirma), mit dem parallel Milliarden von meist ~ 150 DNA-Basen kurzen Sequenz-Schnipseln ausgelesen werden können – also theoretisch ganze Genome auf einmal, nur leider zuvor zu Konfetti zerlegt und ohne weitere Informationen auch nicht mehr vollständig zusammenpuzzelbar. Kombiniert man dieses Verfahren jedoch mit SSR bzw. AFLP, kann man die Schwächen der beiden letzteren zum Teil beheben: Anhand der genauen Sequenzen kann man viel besser sehen, ob es sich tatsächlich um das jeweils selbe Fragment handelt, oder ob sich hinter der scheinbar gleichen Länge verschiedene „Inhalte“ verstecken. Die entsprechenden neuen Verfahren heißen SSRseq (für SSR-Sequenzierung) und RADseq (für „restiction site associated DNA“-Sequenzierung, analog zu AFLP).
Ein Problem bleibt aber auch mit Illumina-Sequenzierung bestehen: Stammen die gelesenen kurzen DNA-Sequenzbereiche tatsächlich vom selben Ort im Genom, oder gibt es sie schlicht mehrfach? Um dies heraus zu bekommen, vergleicht man die RAD- bzw. SSRseq-Daten mit Referenzgenomen, also der vollständigen, nach Chromosomen aufgeteilten DNA-Sequenz eines für die jeweilige Art repräsentativen Individuums. Durchsucht man so ein Referenzgenom, kann man nicht nur sehen ob es Teile davon doppelt gibt, sondern auch neue Mikrosatelliten-Bereiche entdecken oder die Ergebnisse von AFLP- oder RADseq-Studien vorher genauer planen, feststellen in welchen Bereichen des Genoms und wie nahe bei einander die zu vergleichenden Sequenzschnipsel liegen, aus „Genom-Konfetti“ von weiteren Individuen deren +/- komplettes Genom wieder zusammen puzzeln, Gene für bestimmte Eigenschaften der jeweiligen Pflanze heraus finden …
Referenzgenome sind somit zwar enorm nützlich – aber leider für die meisten Arten noch nicht verfügbar. Eines der ersten war das Referenzgenom des Menschen, das von 1997 bis 2006 (mit weiteren Ergänzungen bis 2022) von Forschenden aus mehreren Ländern gemeinsam erstellt wurde. Auf Basis dieses Referenzgenoms konnten viele DNA-Varianten, die bei uns Stoffwechselkrankheiten auslösen oder begünstigen, gefunden und den Betroffenen viel schneller und effektiver geholfen werden. Aber auch „harmlosere“ Informationen, wie etwa Gene, die Haar- und Augenfarbe beeinflussen, wurden teils neu entdeckt – und verhelfen jetzt beispielsweise der Forensik oder der Anthropologie zu neuen Erkenntnissen. Um ähnliches Wissen auch für andere Arten zu schaffen – ganz besonders diejenigen, die aktuell in Gefahr sind, auszusterben – arbeiten zur Zeit weltweit viele Forschende an der Erstellung von weiteren Referenzgenomen. Da dieses Ziel so wichtig ist, haben sie sich zu Arbeitsgemeinschaften wie dem Earth Biogenome Project (EBP – weltweit) oder dem Europäischen Referenz-Genom Atlas (ERGA – Europa und Nachbarländer) zusammen geschlossen. Ziel ist dabei nicht, die Genome der untersuchten Arten zu verändern – sondern sie als Grundlage für ihren effektiven Schutz und evtl. ihre nachhaltige Nutzung im heutigen Zustand zu dokumentieren.
New Technology
Sadly, until this day there is no technology (yet?) through which we can simply read the DNA sequence of an organism while just sitting in front of it. DNA sequencing means first (usually) several days of work in the laboratory, using special machines and chemicals, followed by computer calculations – depending on the amount of data, these days often on a high performance computer. Over time, researchers therefore developed ways to reduce the effort, while at the same time getting more and more accurate and detailed data on potential differences among the genome sequences of individuals.
Mikrosatellites / SSR („short sequence repeats“) are parts of the genome where the very same short sequence motive (e.g. „AC“, „GTT“ or similar) gets repeated multiple times in a cluster (e.g. „ACACACACAC“, „GTTGTTGTT“). When the DNA gets duplicated during cell division, the enzyme responsible for this, called DNA polymerase, often „hiccups“ in these places and gets confused about the copy number. This leads to differences in the number of copies of the sequence motive, or consequently in the length of the whole microsatellite cluster. If the „normal“ sequences before and after the microsatellite cluster are known, it can be selectively copied in the lab and its length can be determined – and compared between different individuals. But this method is prone to some error: variants with the same length / copy number could have originated multiple times and in different ways, other mutations may also have influenced the length of the microsatellite cluster, and sometimes seemingly identical microsatellite clusters exist in the same genome, so that apples might get compared to oranges.
AFLP („amplified fragment lenght polymorphisms“) equally compare just the length, and not the sequence, of certain regions in the genome. To this end, the whole genomic DNA of an individual gets treated with a different type of enzyme, so-called restriction enzymes, which cut the DNA double strand at characteristic sequence motifs (e.g. „A|CCG“). Thereby it is turned into short fragments, and their lengths are subsequently measured. For genetically identical samples, one would expect this to result in an identical list of fragment lengths – otherwise not. But this method suffers from the same shortcomings as the length comparisons in SSR, even more so as the whole genome is involved.
Since a couple of years, there is a new DNA sequencing method (called Illumina sequencing, after its vendor) which can read billions of usually ~ 150 DNA bases long sequence snippets in parallel – so, in theory, whole genomes at once, but unfortunately turned into confetti which cannot be completely puzzled back together without any further information. Yet, if this method is combined with the SSR or AFLP approaches, the weaknesses of the latter two can be partially compensated: With the exact sequences, it is much easier to see if the fragment in question is really the same, or if the apparently identical length hides different „content“. The respective new methods are called SSRseq (SSR sequencing) and RADseq (restriction site associated DNA sequencing, analogous to AFLP).
Still, one problem remains even with Illumina sequencing: Are the short DNA sequence reads originally really from the same spot in the genome, or do they actually exist in multiple places? To find this out, both RADseq and SSRseq data are compared to reference genomes, i.e. the complete DNA sequence, divided up into chromosomes, of an individual representative of the species in question. Searching through such a reference genome, one can not only check if parts of it are duplicated, but also discover new microsatellite clusters or more accurately plan the results of AFLP or RADseq studies, find out in which parts of the genome and how closely together the DNA reads are located, re-assemble +/- the complete genome of further individuals from „genome confetti“ data, find genes for specific properties of the plant being studied …
Reference genomes are thus enormously useful – but not yet available for most species. One of the first was the human reference genome, on which researchers from several countries collaborated from 1997 to 2006 (with further additions until 2022). Based on this reference genome, many DNA variants causing or promoting metabolic diseases in humans were found, and the patients received much faster and more efficient help. But also more „harmless“ information, such as previously unknown genes influencing hair and eye color, was uncovered – and may now help along new discoveries in forensics or anthropology. To reach a comparable level of knowledge for other species – especially those currently under threat of disappearance – many researchers worldwide are currently working on establishing further reference genomes. Because this target is so important, they have come together in collaborative groups such as the Earth Biogenome Project (EBP – worldwide) or the European Reference Genome Atlas (ERGA – Europe and neighbouring countries). The purpose of these efforts is not the modification of these species‘ genomes – but rather, to document them in their current state, as the basis for efficient conservation measures and/or their sustainable use.
Unser Plan
Als Teil des europäischen Projektes Biodiversity Genomics Europe (BGE) gibt unser Arnika-Projekt einen Ausblick auf Naturschutz in einer Welt mit Referenzgenomen: Was wird einfacher, welche Herausforderungen bleiben – und wie können wir diese meistern?
Kosten / Auch wenn die Kosten für DNA-Sequenzierung ständig sinken, ist der Kostenfaktor beim „Sichtbarmachen“ genetischer Vielfalt noch immer der größte Stolperstein. Wir vergleichen daher RADseq- und SSR(seq)-Daten mit dem Arnika-Referenzgenom, um eine Vorstellung zu bekommen, wie repräsentativ diese für die Genom-weite genetische Vielfalt innerhalb der europäischen Arnika sind. Wie sollte ein effizientes, günstiges und möglichst einfaches Verfahren für Genomisches Monitoring aussehen?
Grenzen / Pflanzen halten sich nicht an Ländergrenzen, Naturschutzgesetze gelten aber meist nur innerhalb eines Landes. Auch Messungen der genetischen Vielfalt sind oft auf einzelne Länder beschränkt und die Ergebnisse verschiedener Forschungsgruppen nicht direkt mit einander vergleichbar – dies muss sich ändern. Für Arnika sind wir in der komfortablen Lage, dass bereits mehrere (aber nicht alle) naturschutzgenetischen Arbeitsgruppen, die in Europa an ihr arbeiten, Daten für die gleichen Mikrosatelliten-Bereiche erhoben haben. Darauf möchten wir aufbauen und gemeinsam mit anderen Forschenden herausfinden, worauf man beim Aufbau eines Kontinent-weiten Messsystems für genomische Vielfalt achten muss. Wie kann ein länderübergreifendes System für Genomisches Monitoring aussehen?
Zeit / Gerade weil Arnika eine so bekannte und ikonische Pflanzenart ist, wurde ihre genetische Vielfalt schon lange untersucht. Die ersten Studien dazu stammen bereits aus den 1990er Jahren, sind aber methodisch noch weit von den heutigen Standards entfernt. Aber auch, wenn wir jetzt Referenzgenome haben, ist das kein Grund die alten Daten weg zu werfen – im Gegenteil! Historische Daten zur genetischen Diversität von ausgewählten, auch heute noch genau wiederauffindbaren Pflanzenvorkommen sind ein unbezahlbarer Schatz – und Referenzgenome können uns helfen, diesen zu heben, indem sie es uns erleichtern historische und aktuelle Diversitäts-Daten in Bezug zu setzen. Anhand von Wiederholungsproben einzelner Arnika-Populationen möchten wir testen, wie das Erheben, methodische Abgleichen und Auswerten von genetisch-genomischen Zeitreihendaten funktioniert. Wie kann Genomisches Monitoring evolutionäre Prozesse sichtbar machen?
Wissen / Referenzgenome und Illumina-Sequenzdaten sind super – aber man muss auch mit ihnen umgehen können. Dies wird vor allem für künftige Generationen von Forschenden wichtig sein, die auf unseren heutigen Ergebnissen aufbauen – aber auch andere Menschen sollen verstehen, was wir hier tun und wozu es nützlich ist. Die Mitwirkung von Studierenden, die im Projekt den Umgang mit genomischen Daten lernen und vertiefen, sowie das Einbeziehen und Informieren anderer „Betroffener“, wie etwa lokaler Naturschützer, sind daher ein wichtiger Bestandteil des Projektes. Und auch die wissenschaftsinteressierte Öffentlichkeit soll nicht zu kurz kommen – daher zum Beispiel dieser Blog! Wer wird in Zukunft am Genomischen Monitoring beteiligt sein?
Our plan
As part of the European project Biodiversity Genomics Europe (BGE), our Arnica project gives a preview of conservation in a world with reference genomes: What will get easier, which challenges remain – and how can we master these?
Cost / Even though the cost for DNA sequencing are steadily falling, the money factor remains the greatest stumbling block for „visualising“ genetic diversity. We therefore compare RADseq and SSR(seq) data with the Arnica reference genome, to get an idea of how representative these are for the genome-wide genetic variability within European Arnica. What should an efficient, inexpensive and reasonably simple method for genomic monitoring look like?
Borders / Plants don’t care about borders, but conservation laws usually apply just in individual countries. Moreover, the measurement of genetic diversity is often limited to a single country, and the results of different research groups are not directly comparable – this needs to change. For Arnica, we are in the comfortable position that already several (though not all) conservation genetic research groups working on it in Europe have collected data for the same microsatellite clusters. We want to build further on this and, in collaboration with other researchers, find out what needs to be considered during the establishment of a continental-scale measuring system for genomic diversity. What should an international system for genomic monitoring look like?
Time / Exactly because Arnica is such a well-known and iconic plant species, its genetic diversity has already been surveyed for a long time. The first studies about it date back to the 1990ies, but are methodologically still far away from the current standard. But even if we now have reference genomes, that’s no reason to throw away the old data – on the contrary! Historic data about the genetic diversity of select and today still perfectly re-findable plant populations are a priceless treasure – and reference genomes can help us to uncover it, by making it easier to connect historic and current diversity data. Based on repeat samples of individual Arnica populations, we want to test how the collection, methodological alignment and analysis of genetic-genomic time series data works. How can genomic monitoring make evolutionary processes visible?
Knowledge / Reference genomes and Illumina sequence data are cool – but one needs to know how to handle them. This will be especially important for future generations of researchers, who will build on today’s results – but also other people shall understand what we do and what it is useful for. The participation of students, who will learn and practise to handle genomic data during the project, and the involvement and information of other people „affected“ by it, such as local conservation managers, is therefore an important part of the project. And the science aficionados among the general public shall also get a fair glimpse of the proceedings – this is what this blog is for as well! Who will participate in Genomic Monitoring in the future?
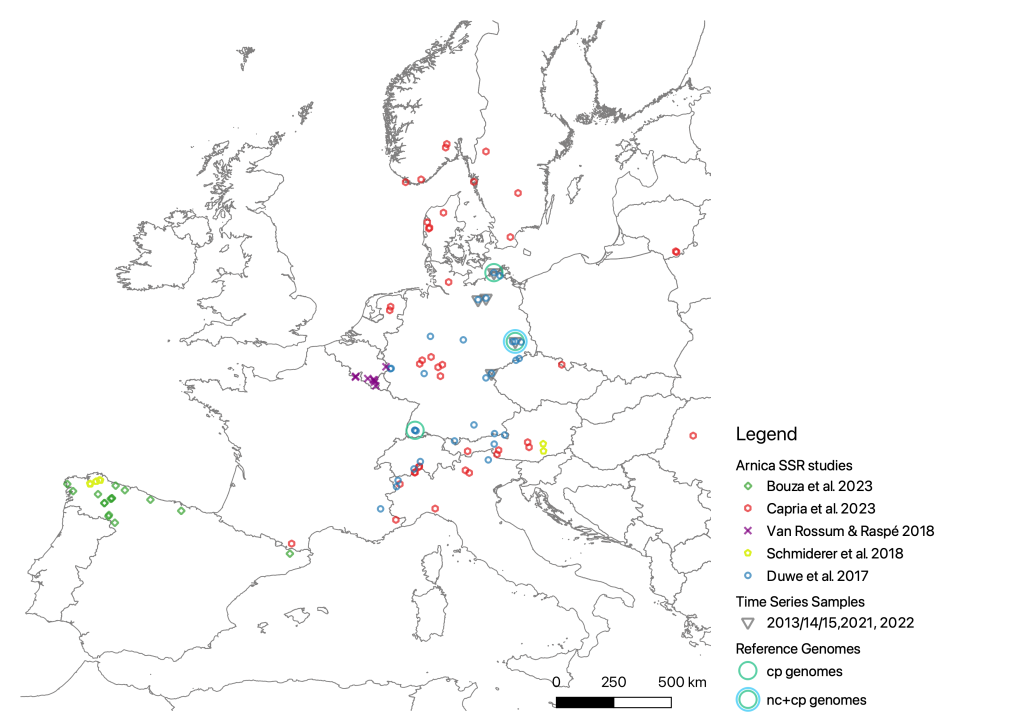
sampling sites of previous conservation genetic Arnica studies. © K. Reichel