
For an upcoming publication, a doctoral student and I want to visualize the sequence variability among several plastid genomes via the tool mVISTA. This tool is often employed in plastid phylogenomic studies and generally simple to use. However, if a user wishes to input custom annotations, it can be quite tricky to generate the correct input for the software. Hence, I automated the generation of the input files as described below.
An effective visualization of plastid genomes in mVISTA requires the x input genomes the user wants to visualize (“input genomes” hereafter; e.g., x=2) as well as a reference genome. For simplicity, a user could employ x1 as the reference genome. The input genomes should be in FASTA format, the reference genome in GenBank format.
To generate custom annotations based on the reference genome, I employ the following Bash code, which (a) converts the reference genome from GenBank format to a cleaned GFF3 format (and incidentally also saves the genome in FASTA format), and (b) converts the cleaned GFF3 file to the mVISTA input.
INF=NC_000932.gb # Just an example
## Converting input file from GenBank format to a cleaned GFF3 format
# Note: This step also generates a FASTA file
grep -vE "codon_start|db_xref|exception" $INF > ${INF}2
to-gff – getfasta ${INF}2 ${INF%.gb*}.gff
grep "gene=" ${INF%.gb*}.gff | \
awk -F';' '{print $1}' | \
grep -vE "remark|intron|misc_feature|repeat_region" > ${INF%.gb*}.gff.clean
rm ${INF}2 ${INF%.gb*}.gff
## Converting input file from GFF3 format to VISTA format
grep -v "^#" ${INF%.gb*}.gff.clean | \
grep -v "rps12" | \
awk '{if ($3 ~ /gene/) {print $7" "$4" "$5" "$3" "$9} else {print $7" "$4" "$5" "$3} }' | \
awk '{if ($4 ~ /gene/) {gsub(/\+/, ">", $1); gsub(/\-/, "<", $1); print $0} else {$1=""; print $0} }' | \
sed 's/gene=//' | \
sed 's/gene/agene/' | \
sort -n -k2 | \
sed 's/agene/gene/' | \
awk '{$1=$1}1' | \
sed 's/CDS/exon/' | \
sed 's/tRNA/utr/' | \
sed 's/rRNA/utr/' > ${INF%.gb*}.mvista
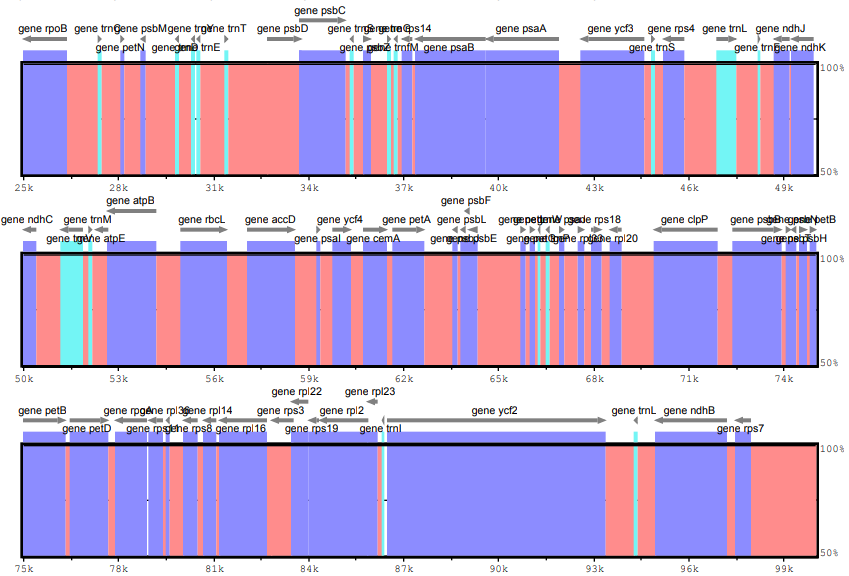
mVISTA example
Am 20. June 2022 um 10:10 Uhr
I was trying to use your script to convert my gb file to gff for mvista format but it didn’t work for me especially this script gives error continuously. what was the case? could you explain me the reason and how should i use please?
Best
Mergi
Am 20. June 2022 um 10:12 Uhr
I was trying to use your script to convert my gb file to gff for mvista format but it didn’t work for me especially this script “to-gff – getfasta ${INF}2 ${INF%.gb*}.gff” gives error continuously. what was the case? could you explain me the reason and how should i use please?
Best
Mergi
Am 29. December 2022 um 01:43 Uhr
I have a similar problem. May I kindly ask you to let me know how I can solve this error?
to-gff: command not found
Am 27. March 2023 um 18:13 Uhr
To anyone getting the error with to-gff, I think this might help:
You have to get the to-gff tool from github:
https://github.com/mscook/to-gff/tree/master
Then you have to install it using python 2, it does not work with python 3.
Since python 2 is deprecated, you can use virtualenv or any other tool to use python 2 instead of python 3.
The line in the example has to be corrected as well, you need to use:
to-gff –getfasta ${INF}2 ${INF%.gb*}.gff
This should solve the problem.
Am 9. August 2023 um 04:38 Uhr
I got the same problem that to-gff: command not found. I substituted this step by converting gb to gff by using NCBI Genome Workbench. Finally, I succeeded in generating the mvista file.
Am 4. February 2024 um 09:23 Uhr
Thank you, Arief. Your method works well.
In simple terms, I converted “.gb” files into “.gff” format using the NCBI Genome Workbench. You can download this software here: https://www.ncbi.nlm.nih.gov/tools/gbench/downloads/
To do this, first, download your target sequences using GenBank accession IDs in NGW and then export your data in “.gff” format.
Secondly, disregard the first part of this script and modify the content as shown below:
## Converting input file from GFF3 format to VISTA format
INF=ID.gb # Just an example
grep -v “^#” “${INF%.gb*}.gff.clean” | \
grep -v “rps12″ | \
awk ‘{if ($3 ~ /gene/) {print $7” “$4” “$5” “$3” “$9} else {print $7” “$4” “$5” “$3} }’ | \
awk ‘{if ($4 ~ /gene/) {gsub(/\+/, “>”, $1); gsub(/\-/, ” “${INF%.gb*}.mvista”
Now you can use the generated outputs from this script to annotate your mvista alignment. Please note that you should manually modify gene names in the generated outputs to align with your gene names above the related locus in the final annotation represented in the output of mvista.
Am 29. February 2024 um 15:37 Uhr
# create new environment and install bcbio-gff
conda create -n mvista
conda activate mvista
conda install bioconda::bcbio-gff
# install github and to-gff
sudo apt install git
git clone https://github.com/mscook/to-gff.git
cd to-gff
pip install .
you should give two hyphen before getfasta. to-gff –getfasta ${INF}2 ${INF%.gb*}.gff