6. October 2020 von Michael Grünstäudl
Recently, a Master’s student of mine asked me to re-calculate some data statistics of an Illumina sequencing run. Among the desired statistics were (a) the total number of read bases (in bp), (b) the total number of reads, (c) the GC content (in %), and (d) the AT content (in %).
Since the average file size of this run was almost a dozen GB per sample, I wrote some Bash code to can calculate these data statistics within minutes.
SAMPLE=mySampleName
#Total read bases (bp):
AllBases=$(grep -A1 "^@" --no-group-separator ${SAMPLE}_*.fastq | \
grep -v "@" | sed "s|$SAMPLE\_.\.fastq-||g" | wc -m)
echo "$AllBases"
#Total reads:
grep "^@" ${SAMPLE}_*.fastq | wc -l
#GC(%):
GandCs=$(grep -A1 "^@" --no-group-separator ${SAMPLE}_*.fastq | \
grep -v "@" | sed "s|$SAMPLE\_.\.fastq-||g" | tr -cd GC | wc -m)
echo "scale=4; $GandCs / $AllBases" | bc
#AT(%):
AandTs=$(grep -A1 "^@" --no-group-separator ${SAMPLE}_*.fastq | \
grep -v "@" | sed "s|$SAMPLE\_.\.fastq-||g" | tr -cd AT | wc -m)
echo "scale=4; $AandTs / $AllBases" | bc
Kategorie bioinformatics, one-liners | 0 Kommentare »
15. September 2020 von Michael Grünstäudl
The process of coding insertions and deletions in a multi-species DNA alignment such that the presence of these indels can be used as additional information in a phylogenetic investigation is typically referred to as “indel coding”. Multiple indel coding schemes exist, with a scheme proposed by Simmons and Ochoterena (2000; often referred to as “simple indel coding scheme”) representing the most widely employed scheme.
Phylogenomic datasets require users to conduct indel coding for dozens, if not hundreds of DNA alignments. However, automating the process of indel coding based on the simple indel coding scheme is fairly straightforward:
We use FHCRC’s Seqmagick to convert NEXUS-formatted alignments to FASTA-formatted alignments, the perl script 2matrix (Salinas and Little 2014) to conduct the indel coding, and then simply conduct some file hygiene afterward.
INF=atpF_intron_aligned_b4manAdj_BETB_exhot.nex
# Convert NEXUS to FASTA
seqmagick convert $INF ${INF%.nex*}.fasta
# Conduct indel coding
perl /path_to_git/2matrix/2matrix.pl -i ${INF%.nex*}.fasta \
-o n,p -n ${INF%.nex*}__SIC
# File hygiene
mv ${INF%.nex*}__SIC.part ${INF%.nex*}__SIC.part.txt
rm ${INF%.nex*}__SIC.garli.nex ${INF%.nex*}__SIC.conf
Kategorie Allgemein | 0 Kommentare »
26. August 2020 von Michael Grünstäudl
The genome assembly process often generates FASTA-formatted contig files, in which the contigs have cryptic sequence names. By using specific Bash commands, one can automatically rename these contigs based on the name of the file they are contained in.
If your contig file contains only a single contig:
for i in *__contig.fasta; do
VAR=${i%__contig.fasta*};
sed -i "1s/.*/>$VAR/" $i;
done
If your contig file contains multiple contigs:
for i in *__contigs.fasta; do
VAR=${i%__contigs.fasta*};
sed -i "s/>.*/>$VAR/" $i;
awk '$0=$0' $i | awk '!(NR%2){print prev "__Length" length($0) ORS $0} {prev=$0}' > ${i}.new
done
Kategorie bioinformatics, one-liners | 0 Kommentare »
8. June 2020 von Michael Grünstäudl
Visualizing the depth
I just published a new paper on an R package that assists users in visualizing the coverage depth of a plastid genome in relation to its circular, quadripartite genome structure. Co-author of the software is an undergraduate student, who (as part of his bachelor thesis) designed the visualization engine of the software.
Kategorie Allgemein | 0 Kommentare »
15. May 2020 von Michael Grünstäudl
Time for innovative methods.
Teaching botany and evolutionary biology to college students during spring 2020 certainly required the application of innovative teaching methods. Classes are held entirely online, and access to suitable plant material is limited by the closure of university greenhouses. Still, with the proper adjustment of the course structure, a meaningful learning experience is possible.

Teaching in May 2020
Kategorie teaching | 0 Kommentare »
31. March 2020 von Michael Grünstäudl
Large-scale DNA sequence submissions to ENA
I just published a new paper on a Python package to submit large DNA sequence datasets to ENA. Special thanks go out to a master’s student of mine who was fantastic in assisting me with testing and debugging the software!
Kategorie Allgemein | 0 Kommentare »
31. January 2020 von Michael Grünstäudl
Teaching.

Teaching in spring 2020
Kategorie teaching | 0 Kommentare »
21. November 2019 von Michael Grünstäudl
High numbers do not equate with high quality
Over the past 10 years, the number of complete plastid genome sequences available on NCBI GenBank has skyrocketed, especially for flowering plants. Whereas in December 2009, approximately 120 complete plastid genomes of flowering plants were present on GenBank, there are 5,838 complete plastid genomes of flowering plants available at the end of November 2019. That is an increase of 4,765% in 10 years (or roughly a quinquaginta-nupling of all plastid genomes available since 2009 [yes, I had Latin in school]).
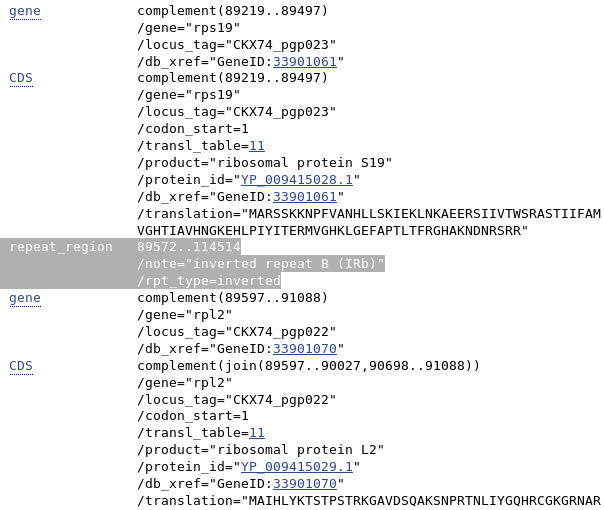
However, it appears that the annotation quality of many of the genomes submitted to GenBank over the past decade remained less than stellar during this increase. The inverted repeat (IR) regions of a plastid genome are integral parts of virtually any flowering plant plastome, and their presence in the sequence annotations are typically a good proxy for the annotation quality. While in December 2009, approximately 95 (or approx. 80%) of the available plastid genomes of flowering plants did not contain unambiguous annotations for the IRs, that number has increased to 3,190 (or approx. 55%) at the end of November 2019.

Example of IR annotation (highlighted in grey) in GenBank flat file of a complete plastid genome
In short, despite a dramatic increase in complete plastid genomes of flowering plants available on GenBank, more than half of all those genomes still do not have appropriate sequence annotations of the IR regions of the plastid genome. This is quite concerning, not least because IR annotations are very simple to include in a genome file (see figure). Clearly, improved strategies for annotating (or correcting) complete plastid genomes are necessary.
Kategorie bioinformatics | 0 Kommentare »
15. October 2019 von Michael Grünstäudl
One of the by-products of being a scientist: Talks at public events

Michael Gruenstaeudl at VictoriaTalk2018
Kategorie myself, teaching | 0 Kommentare »
12. September 2019 von Michael Grünstäudl
Keeping things orderly
Like many other molecular phylogeneticists, I often work with massive numbers of FASTA-formatted DNA sequences. Occasionally, the names of these sequences are numbered in a simplistic fashion (i.e., 1,2,…,48,49), which has the unfortunate side-effect of messing up the intended order of the sequences when sorted numerically, as the sequences 10, 11, …, 19 are followed (and not preceded!) by 1.
To overcome this issue, it is necessary to prepend all sequence names of numbers shorter than the maximum number of digits with one or more leading zeros. The resulting sequence names would, thus, be: 01, 02, …, 48, 49.
Here is a quick Python script that performs the prepending of leading zeros (assuming the FASTA-specific larger-signs are in different lines than the sequence names):
#!/usr/bin/env python2.7
import sys
inFn = sys.argv[1]
delim = '_'
with open(inFn, 'r') as reader, \
open(inFn+'.adjusted', 'w') as writer:
lines = reader.readlines()
for line in lines:
if delim in line and line[0].isdigit():
lparts = [e+delim for e in \
line.split(delim) if e]
lparts[-1] = lparts[-1].rstrip(delim)
if len(lparts[0]) <= 3:
line = lparts[0].rjust(4, '0') + \
''.join(lparts[1:])
writer.write(line)
else:
writer.write(line)
Kategorie bioinformatics | 0 Kommentare »